UGT Inhibition
Understand the potential drug-drug interaction liabilities of your compound by using our UGT inhibition assay.
UGT inhibition is one of Cyprotex's in vitro experimental ADME services. Cyprotex deliver consistent, high quality data with the flexibility to adapt protocols based on specific customer requirements.
Introduction
The inhibition of the UGT enzymes and its significance:
- Uridine glucuronyl transferases (UGT) are a family of enzymes which play a major role in the Phase II metabolism of drugs.
- One in ten of the top two hundred prescribed drugs have glucuronidation as a clearance mechanism illustrating the importance of UGTs in drug metabolism.
- Functionally relevant polymorphisms have been demonstrated for the UGT genes. For example, the polymorphism in UGT1A1 can lead to toxicity associated with Gilbert’s syndrome or the more severe Criglar-Najar syndrome where levels of bilirubin are elevated.
- The regulatory authorities are now recommending that UGT inhibition is evaluated as part of in vitro drug-drug interaction (DDI) packages to determine if clinical DDI studies are required.
- In Cyprotex’s UGT inhibition assay, a decrease in the formation of the UGT-specific metabolite compared to the vehicle control is used to calculate an IC50 value (test compound which produces 50% inhibition). Follow-up Ki determination is also available if required.
- Cyprotex can offer either early stage UGT inhibition screening or regulatory UGT inhibition assessments as part of a DDI package for IND or NDA submissions.
Protocol
UGT Inhibition Assay Protocol
Data
Data from Cyprotex's UGT Inhibition Assay
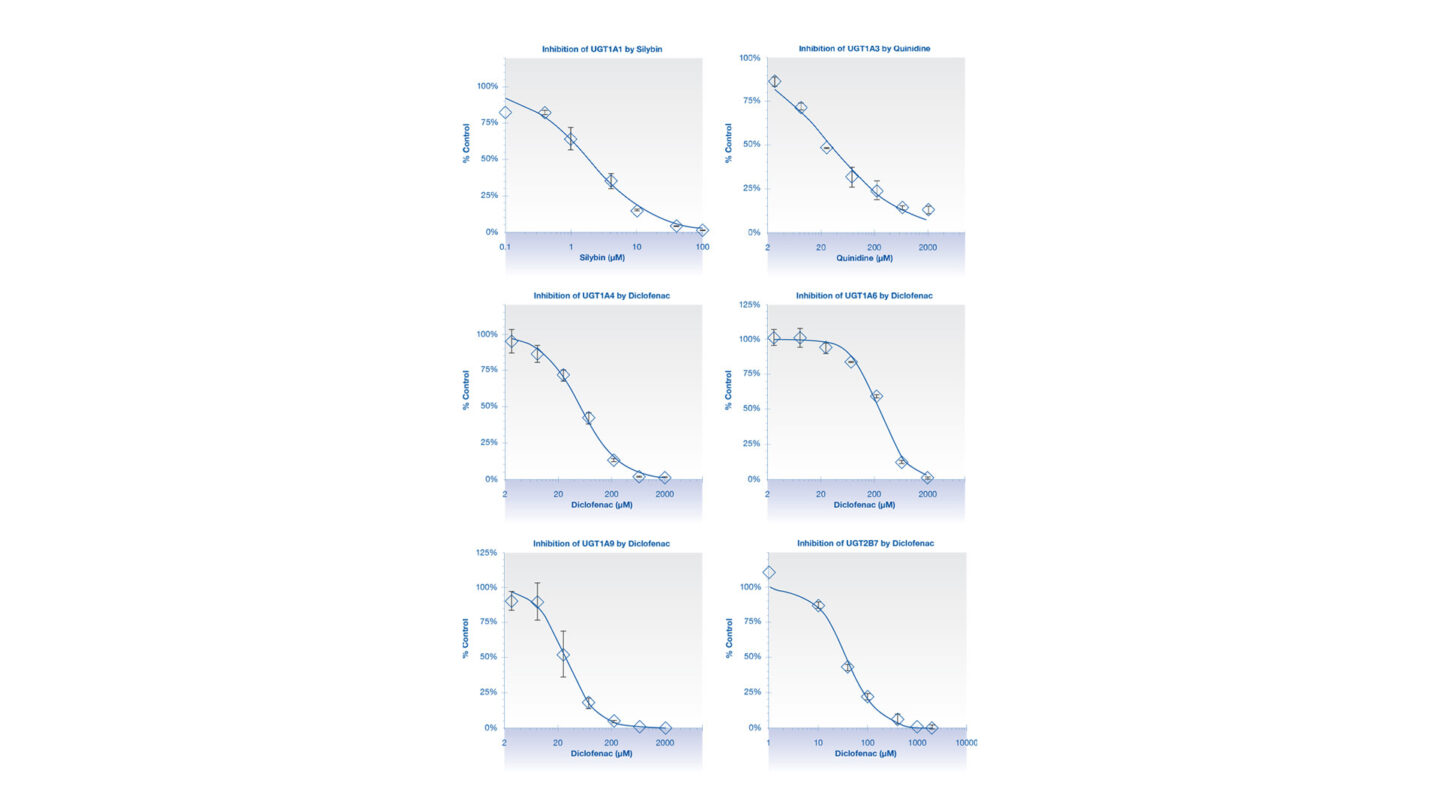
Figure 1
Graphs showing the inhibition of UGT isoforms by the positive control inhibitors in Cyprotex’s UGT inhibition assay. Data show the mean ± standard deviation of 3 replicates.
Q&A
Why is glucuronidation an important clearance mechanism?
Uridine glucuronyl transferases (UGT) are a family of enzymes which play a major role in the metabolism of drugs. The importance of glucuronidation is highlighted by the fact that it is a listed clearance mechanism for 1 in 10 of the top 200 prescribed drugs2.
Glucuronidation involves the addition (or conjugation) of glucuronic acid either directly to the drug itself or to an oxidative metabolite of the drug. The addition of a glucuronide results in conjugates that are more polar and ionized at physiological pH. These features facilitate excretion via the kidneys. Glucuronides may also be excreted by the liver through the bile.
Why is it important to investigate UGT inhibition?
UGT1A1 is an important isoform of the UGT family of enzymes due to its role in the glucuronidation of the endogenous product, bilirubin, as well as estrogens and flavanoids. Certain individuals also exhibit a polymorphism in this isoform which may impair their ability to metabolize certain drugs. Inhibition of UGT1A1 enzyme has the potential to produce increased levels of bilirubin in the circulation which can lead to toxicological effects. Drug-drug interactions may be of greater significance in individuals who are carriers of the polymorphic variant of UGT1A1.
Other members of the UGT family (in addition to UGT1A1) are now becoming of interest due to their role in drug metabolism and their potential for clinical drug-drug interactions. These include UGT1A3, UGT1A4, UGT1A6, UGT1A9 and UGT2B7. The regulatory authorities are starting to request these studies as part of drug-drug interaction (DDI) packages to determine if clinical DDI studies are required.
It is important to note that UGT clinical DDI’s are much less likely than with CYP enzymes. This is because UGT substrates are often metabolized by multiple UGTs and have high Km values compared to CYP substrates.
Please provide an overview of the UGT inhibition assay.
For the UGT inhibition assay, the known UGT substrate is incubated with cDNA-expressed human UGT-specific Supersomes™, alamethicin, UDPGA and a range of test compound concentrations at 37°C. The protein concentration and time point is dependent on the particular UGT and has been previously determined by protein linearity and time linearity studies. The substrate concentration has previously been determined by characterizing the enzyme kinetics of each reaction. The range of test compound concentrations is dependent on the plasma Cmax, the extent of plasma protein binding and the solubility of the test compound.
At the end of the incubation, the formation of the UGT isoform-specific metabolite is monitored by LC-MS/MS at each of the test compound concentrations. A decrease in the formation of the metabolites compared to vehicle control is used to calculate an IC50 value (test compound concentration which produces 50% inhibition).
What substrates are used in the UGT inhibition assay?
The following substrates are used for the UGT inhibition assays:
- UGT1A1: Estradiol
- UGT1A3: Sulindac sulfone
- UGT1A4: Trifluoperazine
- UGT1A6: Naphthol
- UGT1A9: Propofol
- UGT2B7: Naloxone
What positive control inhibitors are used in the UGT inhibition assays?
The following positive control inhibitors are used in the UGT inhibition assays:
- UGT1A1 inhibitor: Silybin or atazanavir
- UGT1A3 inhibitor: Quinidine
- UGT1A4, UGT1A6, UGT1A9 and UGT2B7 inhibitor: Diclofenac
What is the concentration of the probe substrates relative to Km?
The substrate concentration is approximately equivalent to the Km.
Why do you include UDPGA and alamethicin in the incubation?
UDPGA (UDP-glucuronic acid) is the conjugating agent required for formation of the glucuronide. It is necessary to supplement with this cofactor for glucuronidation to occur. Due to the luminal location of the UGT's within the endoplasmic reticulum of microsomal preparations, the passage of the water soluble cofactor UDPGA to the active site is challenging. In order to overcome this latency phenomenon, the pore forming agent, alamethicin, can be used to improve access to the active site. Supersomes have a lower level of latency than mammalian tissue fractions.
Why do you use expressed enzyme rather than human liver microsomes to study UGT inhibition?
Human liver microsomes contain numerous Phase I and Phase II enzymes. Substrate specificity of UGTs is not as well defined as compared to the CYP enzymes. For example, although UGT1A1 is the main isoform involved in the 3β-glucuronidation of estradiol, it has been shown, based on relative activity data, that UGT1A3 may account for up to one third of the formation of estradiol 3β -glucuronide3. Additionally, UGT2B7 is responsible for the formation of estradiol 17β-glucuronide4. By using expressed enzyme, the metabolism of the substrate is not being complicated by any other metabolic reaction and is solely focusing on the UGT being evaluated.
Can I further characterise the type of inhibition determined?
Following IC50 determination we can then determine the Ki for the test compound against the appropriate isoform. This will give information as to the potency of the inhibition and the type of inhibition (competitive, non-competitive, uncompetitive or mixed) and can be used to estimate the impact of any potential in vivo interactions.
References
1) Remmel R et al., (2007) Conjugative Metabolism of Drugs, in Drug Metabolism in Drug Design and Development, (Zhang D et al.eds); 37-88, John Wiley & Sons, Inc.
2) Williams JA et al., (2004) Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCI/AUC) ratios. Drug Metab Dispos 32(11); 1201-1208
3) Miners JO et al., (2010) The prediction of drug-glucuronidation parameters in humans: UDP-glucuronosyltransferase enzyme-selective substrate and inhibitor probes for reaction phenotyping and in vitro-in vivo extrapolation of drug clearance and drug-drug interaction potential. Drug Metab Reviews 42(1); 196-208
4) Gall WE et al., (1999) Differential glucuronidation of bile acids, androgens and estrogens by human UGT1A3 and 2B7. J Steroid Biochem Mol Biol 70; 101-108
Downloads
Contact Our Experts