Cytochrome P450 (CYP) Inhibition assay (IC50)
Understand the potential drug-drug interaction liabilities of your compounds by using our cytochrome P450 (CYP450) reversible inhibition assay for a range of isoforms.
The cytochrome P450 inhibition assay is one of our portfolio of in vitro ADME screening services. Cyprotex delivers consistent, high quality data with cost-efficiency that comes from a highly automated approach.
Introduction
Inhibition of cytochrome P450 (CYP) enzymes:
- CYP's are a family of enzymes which play a major role in the metabolism of drugs.
- Assessment of the potential of a compound to inhibit a specific CYP enzyme is important as co-administration of compounds may result in one or both inhibiting the other’s metabolism. This may affect plasma levels in vivo and potentially lead to adverse drug reactions or toxicity.
- In vitro CYP inhibition data are useful in designing strategies for investigating clinical DDI Studies.
- Cyprotex's cytochrome P450 inhibition assays use industry accepted probe substrates and human liver microsomes.
- In Cyprotex's cytochrome P450 inhibition assay, a decrease in the formation of the metabolites compared to the vehicle control is used to calculate an IC50 value (test compound concentration which produces 50% inhibition).
Protocol
Cytochrome P450 Inhibition Assay Protocol (IC50)
Data
Data from Cyprotex's Cytochrome P450 Inhibition Assay
Known CYP inhibitors were screened in Cyprotex's Cytochrome P450 Inhibition assay in 3 separate assays.
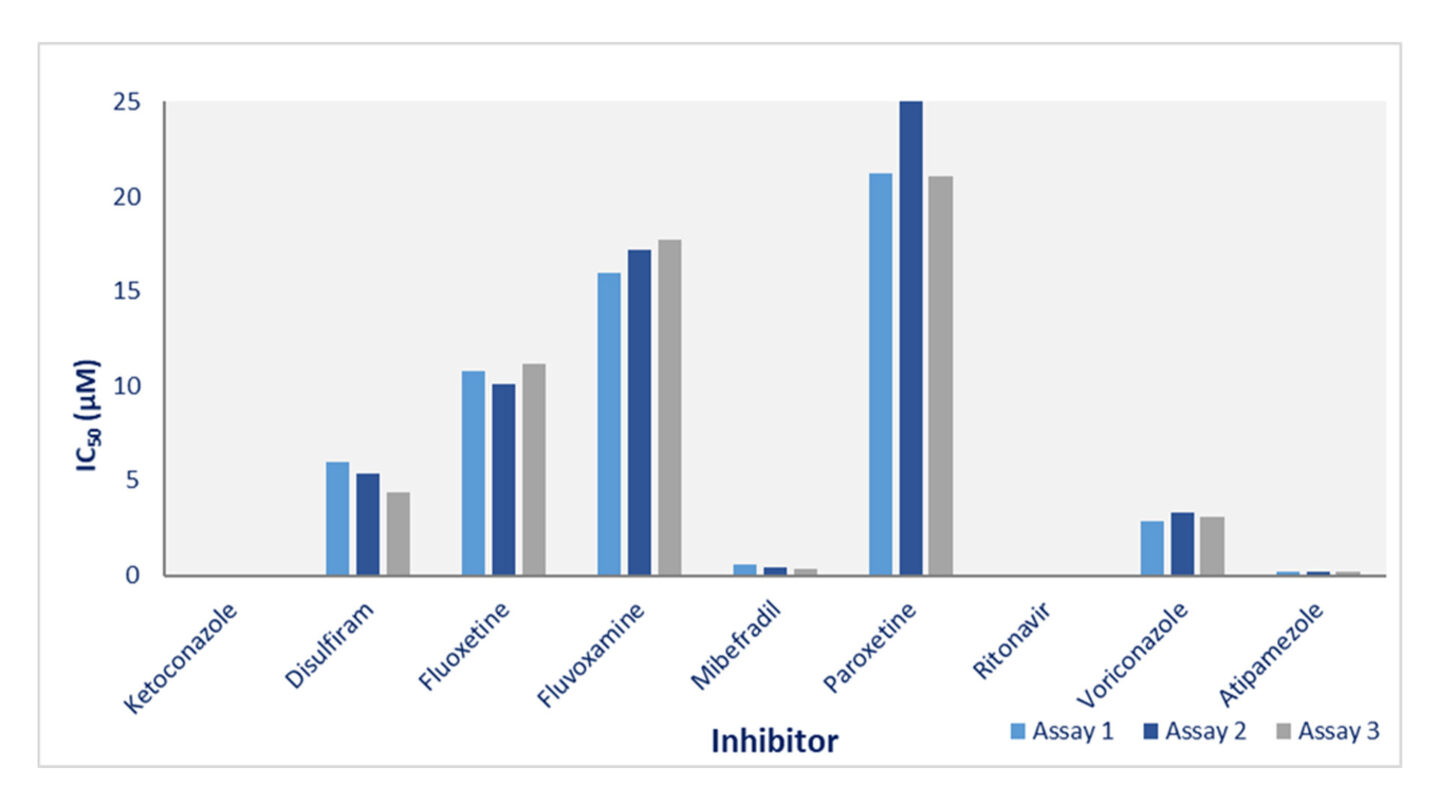
Figure 1
Cyprotex's cytochrome P450 inhibition data for CYP3A4. The effect of 8 potential inhibitors (disulfiram, fluoxetine, fluvoxamine, mibefradil, paroxetine, ritonavir, voriconazole and atipamazole) and known CYP3A4 inhibitor ketoconazole on the 1-hydroxylation of midazolam was investigated on 3 separate occasions. The data show good consistency for inhibitors with a range of inhibition potential.
Q&A
Please provide an overview of the Cyprotex's Cytochrome P450 Inhibition assay.
Reversible inhibition can be investigated for the seven main CYP isoforms (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4) with two substrates recommended for CYP3A46.. Isoform-specific substrates are incubated individually with a range of test compound concentrations (typically 0.1 - 25 µM) in the presence of human liver microsomes and cofactor. At the end of the incubation, the formation of metabolite is monitored by LC-MS/MS
A decrease in the formation of the metabolites compared to vehicle control is used to calculate an IC50 value (test compound concentration which inhibits enzyme activity by 50%).
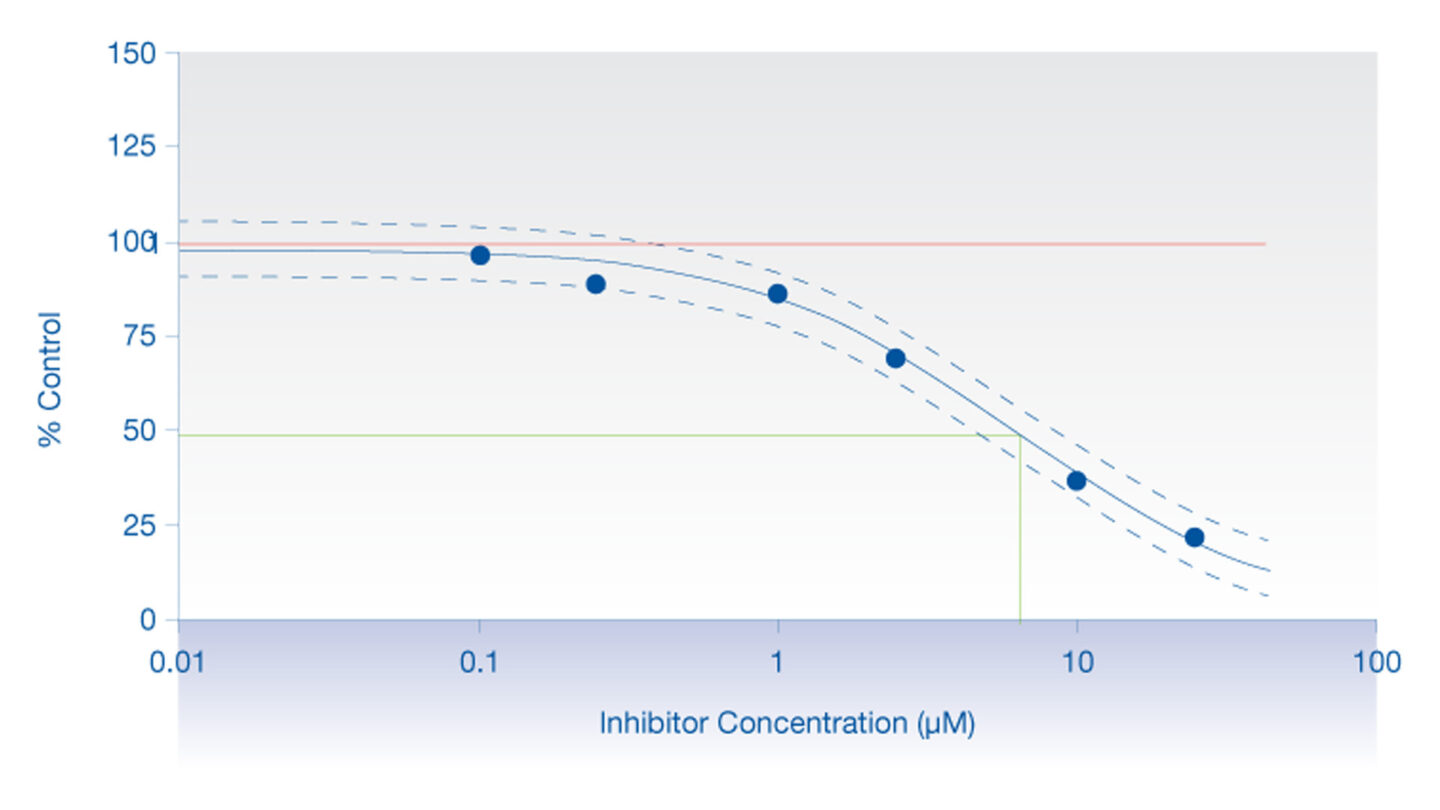
Figure 2
Graph displaying a typical inhibition profile.
Why is it important to investigate cytochrome P450 inhibition?
Evaluating the potential CYP inhibition of a new investigational drug is recommended in the FDA guidance for drug interaction studies (2020)6 and the EMA guideline on the investigation of drug interactions (adopted 2012)7. The CYP enzymes play a major role in the metabolism of the majority of drugs and, because of the high specificity and low capacity of many of these enzymes, they are most likely to be involved in clinically relevant DDI. Quantitative assessment of the in vitro data in conjunction with clinical pharmacokinetic data is used to determine if an in vivo clinical DDI study is required. It is important to note that major metabolites should also be investigated for drug interaction potential.
How do I interpret the data from the cytochrome P450 (CYP) inhibition assay?
Inhibition potency must always be considered in the context of expected in vivo concentrations of the test compound. Although the criteria for acceptance are project and isoform-specific, potent inhibition is considered unfavourable and may preclude the development of a compound.
What is the concentration of the probe substrates relative to Km?
The substrate concentration is equivalent to the Km.
Do you perform the assays for each isoform as a cocktail, or as separate incubations?
All of our CYP inhibition reactions are incubated separately for each isoform.
Why do you use human liver microsomes rather than cDNA expressed enzyme to study P450 inhibition?
Metabolizing systems which use recombinant enzymes are artificial as the enzyme is not present in its native environment and is often over-expressed. With these systems, there is an absence of competing enzymes and reactions. Human liver microsomes contain the full complement of CYP enzymes and so are more comparable to the in vivo situation.
Can I further characterise the type of inhibition determined?
Following IC50 determination we can then determine the Ki for the test compound against the appropriate isoform. This will give information as to the potency of the inhibition and the type of inhibition (competitive, non-competitive, uncompetitive or mixed) and can be used to estimate the impact of any potential in vivo interactions.
References
1) Obach RS et al. (2006) JPET 316; 336-348
2) Perloff et al., (2009), Xenobiotica, 39(2):99-112
3) Hesse et al., (2000) DMD, 28(10):1176-1183
4) Walsky et al., (2005) DMD, 33(3):413-418
5) Walsky et al., (2005) DMD 32(6):647-660
6) US FDA guidance for industry - In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions (2020)
7) The European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions (Adopted in 2012)
Downloads
Contact Our Experts