Cytochrome P450 Time Dependent Inhibition (IC50 shift) Assay
Understand the potential drug-drug interaction liabilities of your compounds by using our cytochrome P450 (CYP450) time dependent inhibition assays for a range of isoforms.
Our range of cytochrome P450 time dependent inhibition services form part of our portfolio of in vitro ADME screening services. Cyprotex delivers consistent, high quality data with cost-efficiency that comes from a highly automated approach.
Introduction
Time dependent inhibition of cytochrome P450 (CYP450) enzymes:
- Inhibition of cytochrome P450 enzymes is one of the most common mechanisms resulting in clinically relevant drug-drug interactions. This inhibitory effect can either be a reversible or irreversible (time dependent) interaction.
- Time dependent inhibition (TDI) of cytochrome P450 is of particular concern as typically de novo synthesis of the enzyme is required in order to restore activity. The consequences of TDI can be termination of drug development, drug withdrawal or serious restrictions of use.
- Cyprotex’s IC50 shift assay determines the IC50 (inhibitor concentration which results in 50% inhibition of activity) following a pre-incubation in the absence and presence of NADPH. This assay enables discrimination between compounds which cause reversible, irreversible, or both reversible and irreversible inhibition.
Related Services:
Protocol
Cytochrome P450 Time Dependent Inhibition (IC50 Shift) Assay Protocol
Data
Data from Cyprotex's Cytochrome P450 Time Dependent Inhibition (IC50 Shift) assay
A number of known time dependent inhibitors were screened in the IC50 shift assay in triplicate. Inhibitors which were known to be solely reversible inhibitors were screened alongside the time dependent inhibitors as negative controls. The results show a high level of consistency over a range of inhibition values.
Q&A
Please provide an overview of the cytochrome P450 time dependent inhibition (IC50 shift assay).
The Cytochrome P450 Time Dependent Inhibition IC50 Shift assay is able to identify both reversible and time-dependent inhibitors. The IC50 shift assay determines the IC50 value (concentration which produces 50% inhibition) of test compound under three different experimental conditions; 0 min pre-incubation, 30 min pre-incubation minus NADPH and 30 min pre-incubation plus NADPH. Following the pre-incubation with/without NADPH, isoform-specific substrates are added to measure the residual enzyme activity without a dilution step, running the incubation under linear conditions. If the compound is a time-dependent inhibitor a shift to the left (increase in potency) will occur between the 30 min pre-incubation minus NADPH and 30 min pre-incubation plus NADPH. The ratio of these two values gives the IC50 shift.
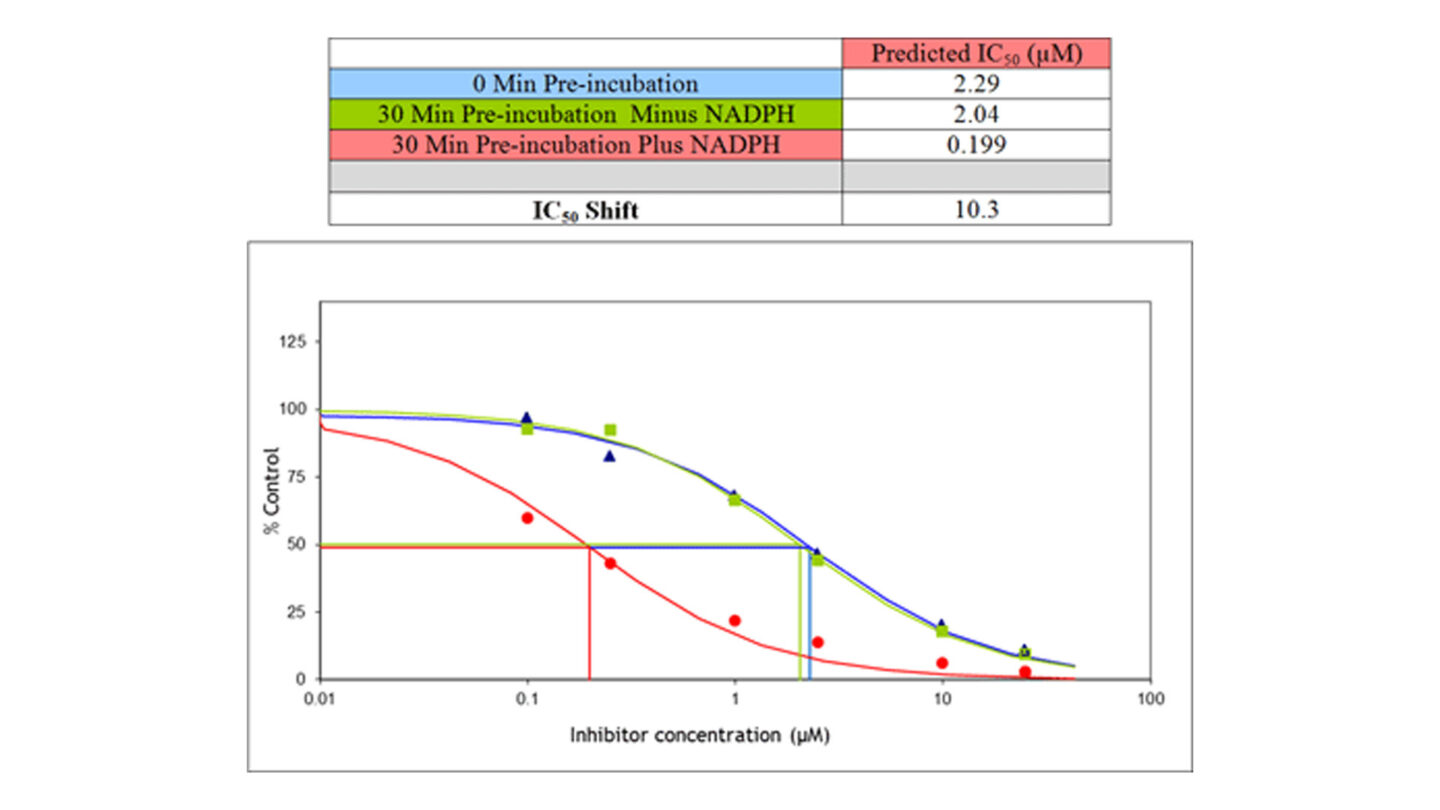
Figure 4
Example IC50 shift profile
Why is it important to evaluate time dependent inhibition?
Cytochromes P450 (CYP) are a family of enzymes which play a major role in the metabolism of drugs. Inhibition of CYP enzymes is one of the most common causes of drug-drug interactions. The mechanism of inhibition can be reversible, quasi-irreversible or irreversible.
The consequences of irreversible inhibition are considered to be more serious than reversible inhibition because the inactivated enzyme must be re-synthesized before activity is restored. In addition, the irreversible inactivation usually implies the formation of a covalent bond between the metabolite and the enzyme, which can lead to hapten formation and can in some cases trigger an autoimmune response. For these reasons it is important to study the mechanism of the CYP inhibition of new potential drugs as early as possible during the drug discovery process.
Although the terms time dependent inhibition (TDI) and mechanism based inhibition (MBI) are often used inter-changeably, there is a distinct difference scientifically. Time dependent inhibition is defined as an interaction where there is an enhanced inhibition if the test compound is pre-incubated with the metabolizing system prior to addition of the substrate. Mechanism based inhibition specifically refers to a subset of time dependent inhibition which defines inactivation of the enzyme by a chemically reactive metabolite1.
The FDA guidance for drug interactions (January 2020)2 recommend evaluating time dependent inhibition for investigational drugs.
How do I interpret the data from the IC50 shift assay?
Using the IC50 shift assay, it is possible to distinguish between reversible and irreversible inhibition. For solely reversible inhibitors addition of a 30 min pre-incubation gives no change in the IC50. Consequently all three experimental conditions will give the same IC50. Alternatively for compounds which are only time dependent inhibitors, an IC50 will be calculated only when NADPH is present in the pre-incubation and no effect is observed in the 0 min pre-incubation or 30 min pre-incubation minus NADPH. Test compounds which exhibit both reversible and time dependent inhibition will give a lower IC50 with a 30 min pre-incubation in the presence of NADPH compared to a 30 min pre-incubation minus NADPH and 0 min pre-incubation and thus an IC50 shift can be calculated as shown in Figure 4.
Very occasionally a shift may be observed between the 0 min pre-incubation and 30 min pre-incubation minus NADPH. This indicates potential non-NADPH mediated metabolism of one test compound into a more potent inhibitor species although this is rarely observed with CYP450 enzymes3.
A fold shift of greater than 1.5 is considered to be a significant shift and the compound is classed as a time dependent inhibitor4. Inhibition potency must always be considered in the context of expected in vivo concentrations of the test compound. Although the criteria for acceptance are project and isoform-specific, potent inhibition is considered unfavourable and may preclude the development of a compound. Further work should then be completed to evaluate the mechanism of time-dependent inhibition or determine KI and kinact values to predict the risk of drug-drug interactions in vivo5.
Why is there no dilution step between the pre-incubation and incubation steps?
A dilution step is an additional factor in the assay conditions which can skew the behavior of the inhibitor and complicate data processing. Pre-incubating the test compound at 10x higher microsomal protein concentration than that in the incubation step introduces the possibility for higher protein binding, reducing the amount of free compound available for inhibition. This can give an under-prediction of potency. Additionally, differences can be observed in data processing depending on whether the inhibitor concentration in the ‘pre-incubation’ is taken, compared with the ‘incubation’ i.e. diluted 1 in 10. Thus could give a difference in potency of 10×. The effect is more pronounced upon compounds which have a reversible inhibition element compared with those which are solely time-dependent inhibitors.
Consequently an IC50 shift assay which does not have a
dilution step gives the most accurate representation of a 30 min
pre-incubation, providing unambiguous information for analyzing
compounds for further work.
Can I further characterize the time-dependent inhibition determined?
If an IC50 shift is observed, a KI and kinact determination assay should be determined. This ascertains the time-dependent inhibition kinetic constants, where kinact is the maximal rate of enzyme inactivation at a saturating concentration of inhibitor and KI is the concentration of inhibitor which gives half the maximal rate of inactivation. Alongside in vivo data, this can be used to predict the risk of drug-drug interactions (AUCi/AUC)5.
References
1) Grimm SW et al. (2009) Drug Metab Dispos 37; 1355-1370
2) FDA Guidance for Industry – In Vitro Drug Interaction Studies - Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions (January 2020)
3) Wright JN et al. (1991) Biochem J 273(pt3); 533-539
4) Berry LM et al. (2008) Drug Metab Lett 2; 51-59
5) Burt HJ et al. (2010) Xenobiotica 40(5); 331-343
Downloads
Contact Our Experts